This web page was produced as an assignment for genetics 564, an undergraduate course at UW-Madison
Domains and Motifs
What are domains and motifs?
Motifs are defined as conserved regions of spatially close sequences (5,6). Multiple motifs can comprise a domain. A domain has 3D structures associated with it as well as an associated function (6). These similar functional units can be conserved and found in a variety of different proteins. Due to the fact many proteins possess the same associated stretch of amino acids that give rise to these conserved domains, databases can predict the domains found in a protein sequence. Moreover, upon inputting a protein sequence in the database, the database is able to find conserved regions and predict the activities associated with the protein sequence. Domains typically are made up of about 50 amino acid residues (6). Domains are usually involved with protein-protein interactions. A single protein can have one or multiple domains. Using various databases, researchers determined the domains in ACVR1 to give insight into information about the functions and activities of this protein.
How do you find protein domains and motifs?
Using PFAM and SMART, the domains and motifs can be identified within the activin A receptor protein (1,3). On PFAM, by plugging in the FASTA format of the protein sequence, these databases use algorithms to predict likely domains and motifs. The output of PFAM is shown in image one. This demonstrates that researchers identified three domains and the locations of these domains within the peptide sequence, namely the protein kinase domain, TGF-beta motif and activin receptor domain. These databases were used to look at the domains and motifs present in a variety of homologs to the human ACVR1 protein. The results are listed below in image two.
What are domains and motifs?
Motifs are defined as conserved regions of spatially close sequences (5,6). Multiple motifs can comprise a domain. A domain has 3D structures associated with it as well as an associated function (6). These similar functional units can be conserved and found in a variety of different proteins. Due to the fact many proteins possess the same associated stretch of amino acids that give rise to these conserved domains, databases can predict the domains found in a protein sequence. Moreover, upon inputting a protein sequence in the database, the database is able to find conserved regions and predict the activities associated with the protein sequence. Domains typically are made up of about 50 amino acid residues (6). Domains are usually involved with protein-protein interactions. A single protein can have one or multiple domains. Using various databases, researchers determined the domains in ACVR1 to give insight into information about the functions and activities of this protein.
How do you find protein domains and motifs?
Using PFAM and SMART, the domains and motifs can be identified within the activin A receptor protein (1,3). On PFAM, by plugging in the FASTA format of the protein sequence, these databases use algorithms to predict likely domains and motifs. The output of PFAM is shown in image one. This demonstrates that researchers identified three domains and the locations of these domains within the peptide sequence, namely the protein kinase domain, TGF-beta motif and activin receptor domain. These databases were used to look at the domains and motifs present in a variety of homologs to the human ACVR1 protein. The results are listed below in image two.

Image 1: This illustrates the domains and motifs derived from using PFAM for ACVR1 in humans.
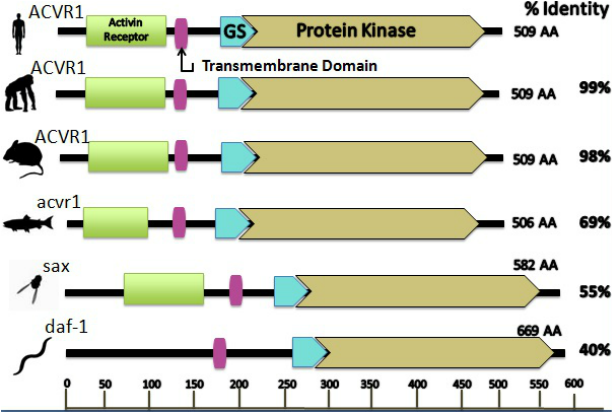
Image 2: This illustrates the protein domains and motifs present in a variety of different model organisms.
Analysis:
From comparing the motifs and domains derived from the databases SMART and PFAM, it can be seen that the predicted protein domains and motifs are nearly identical. The exception is with SMART. SMART possesses an additional transmembrane domain. Both of all these databases use different algorithms which may account for the slightly different results. By combining the information found from both databases, the final predicted protein and associated domains are illustrated in image two. The domains that were embedded in the protein sequence include the activin receptor domain, a transmembrane domain, TGF-beta motif and a protein kinase domain. These domains coincide with what would be expected considering what is understood about the mechanism of the disease. Another important feature of the protein is that these domains and motifs are highly conserved in vertebrates as well as some invertebrates. However, there is an absence of the activin receptor domain in the C. elegan. This may be accounted for by the fact this protein is only 40 percent identical to the human homolog. Additionally, these species are invertebrates and may not require the same bone formation pathways that are crucial to vertebrates. The first domain is the activin receptor domain. Considering the protein that is encoded by the gene ACVR1 is the activin A receptor type I, it makes sense that the protein would consist of the activin receptor domain for which it is named. Secondly, SMART predicted a transmembrane domain present in the protein. From gene ontology and the literature, these resources illustrate that the protein is localized in the membrane of a cell. Thus, having a transmembrane domain would allow for this localization. Additionally, PFAM and SMART predicted the presence of the motif for transforming growth factor beta (TGF-beta). The mutation resides in one of the amino acids comprising this motif. This is essential and can explain the disease phenotype and mechanism. This motif is crucial for regulating cell growth and differentiation by binding the associated ligand and inducing the catalytic activity of the receptor to undergo signaling events that correspond to gene expression changes of bone associated genes. In soft tissue, this pathway is prohibited by a molecule of called FKBP12, which binds to the TGF-beta GS region and prevents singling of the bone morphogenetic pathway (8,9,10). In the pathological state, the single mutation alters the activity of the TGF-beta domain by making the inhibitor unable to bind to this motif. Consequently, this signaling is constitutively active and induces bone gene expression changes that result in the cell differentiating into bone (8,9,10). The expression of these bone formation genes is what induces the soft tissue cells to differentiate into bone cells. PFAM indicates that “point mutations in GS modify signaling of the receptor,” which is exactly what is observed in the disease. This motif is present in many species and is highly conserved. The last domain is protein kinase. These domains allow for the receptor to have catalytic activity, meaning the receptor acts as an enzyme that can phosphorylate other proteins and cause an abundance of downstream cellular changes, namely, metabolism, transcription, cell cycle progression, cytoskeletal rearrangement, cell movement, apoptosis and differentiation. Due to the vast amount of cellular functions of this domain, it is highly conserved. By analyzing the protein domains, this can provide indications or verify a proteins activity.
From comparing the motifs and domains derived from the databases SMART and PFAM, it can be seen that the predicted protein domains and motifs are nearly identical. The exception is with SMART. SMART possesses an additional transmembrane domain. Both of all these databases use different algorithms which may account for the slightly different results. By combining the information found from both databases, the final predicted protein and associated domains are illustrated in image two. The domains that were embedded in the protein sequence include the activin receptor domain, a transmembrane domain, TGF-beta motif and a protein kinase domain. These domains coincide with what would be expected considering what is understood about the mechanism of the disease. Another important feature of the protein is that these domains and motifs are highly conserved in vertebrates as well as some invertebrates. However, there is an absence of the activin receptor domain in the C. elegan. This may be accounted for by the fact this protein is only 40 percent identical to the human homolog. Additionally, these species are invertebrates and may not require the same bone formation pathways that are crucial to vertebrates. The first domain is the activin receptor domain. Considering the protein that is encoded by the gene ACVR1 is the activin A receptor type I, it makes sense that the protein would consist of the activin receptor domain for which it is named. Secondly, SMART predicted a transmembrane domain present in the protein. From gene ontology and the literature, these resources illustrate that the protein is localized in the membrane of a cell. Thus, having a transmembrane domain would allow for this localization. Additionally, PFAM and SMART predicted the presence of the motif for transforming growth factor beta (TGF-beta). The mutation resides in one of the amino acids comprising this motif. This is essential and can explain the disease phenotype and mechanism. This motif is crucial for regulating cell growth and differentiation by binding the associated ligand and inducing the catalytic activity of the receptor to undergo signaling events that correspond to gene expression changes of bone associated genes. In soft tissue, this pathway is prohibited by a molecule of called FKBP12, which binds to the TGF-beta GS region and prevents singling of the bone morphogenetic pathway (8,9,10). In the pathological state, the single mutation alters the activity of the TGF-beta domain by making the inhibitor unable to bind to this motif. Consequently, this signaling is constitutively active and induces bone gene expression changes that result in the cell differentiating into bone (8,9,10). The expression of these bone formation genes is what induces the soft tissue cells to differentiate into bone cells. PFAM indicates that “point mutations in GS modify signaling of the receptor,” which is exactly what is observed in the disease. This motif is present in many species and is highly conserved. The last domain is protein kinase. These domains allow for the receptor to have catalytic activity, meaning the receptor acts as an enzyme that can phosphorylate other proteins and cause an abundance of downstream cellular changes, namely, metabolism, transcription, cell cycle progression, cytoskeletal rearrangement, cell movement, apoptosis and differentiation. Due to the vast amount of cellular functions of this domain, it is highly conserved. By analyzing the protein domains, this can provide indications or verify a proteins activity.
Protein Interactions
What are protein interactions?
Proteins are the functional components controlling the biological systems in a cell. Proteins occasionally perform functions independently; however, more commonly, proteins function in a complex interaction with multiple proteins. These networks with a protein of interest are called protein interactions. By investigating these protein interactions in connection with a protein of interest can provide clues into the biological function of those proteins in the cell. Various databases can provide information about protein interactions and networks in a cell such as STRING, IntAct, BioGRID.
What proteins are interacting with ACVR1?
Using STRING, the protein interactions of ACVR1 were investigated. The results are shown below in Figure 3. As shown in Figure 4, the primary function of the proteins in this pathway appear to be in transcriptional regulation, bone induction, transmembrane receptors, and inhibitors to protein kinase receptors. Therefore, it is not surprising that the stimulation of these proteins in the disease state would result in the excessive bone growth phenotype seen in patients. Therefore, these results coincide with the literature and disease phenotype.
What are protein interactions?
Proteins are the functional components controlling the biological systems in a cell. Proteins occasionally perform functions independently; however, more commonly, proteins function in a complex interaction with multiple proteins. These networks with a protein of interest are called protein interactions. By investigating these protein interactions in connection with a protein of interest can provide clues into the biological function of those proteins in the cell. Various databases can provide information about protein interactions and networks in a cell such as STRING, IntAct, BioGRID.
What proteins are interacting with ACVR1?
Using STRING, the protein interactions of ACVR1 were investigated. The results are shown below in Figure 3. As shown in Figure 4, the primary function of the proteins in this pathway appear to be in transcriptional regulation, bone induction, transmembrane receptors, and inhibitors to protein kinase receptors. Therefore, it is not surprising that the stimulation of these proteins in the disease state would result in the excessive bone growth phenotype seen in patients. Therefore, these results coincide with the literature and disease phenotype.
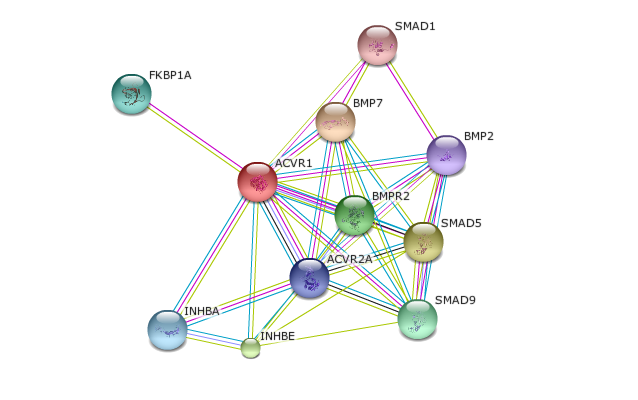
Figure 3: This image shows the network of protein interactions involved with the disease causing protein, ACVR1.

Figure 4: This image summarizes the activities of the proteins that interact with ACVR1
Analysis:
Studies have confirmed that a missense mutation the 206th amino acid in the ACVR1 protein is consistently correlated with FOP. Research has also confirmed that ACVR1 is a transmembrane protein that receives signals from the outside of the cell and facilitates internal cellular changes that ultimately alter gene expression. Therefore, from investigating the information derived from databases like STRING, these interactions confirm what is known from the literature. Using other databases such as PFAM and SMART, domains in ACVR1 were identified and conclusions into protein activity can be inferred from the outputs. As mentioned above, these databases predicted that ACVR1 has kinase ability which confers the protein to stimulate cell signaling upon ligand binding. This information is also present in the literature. Therefore, activation of this protein has enzymatic activity to initiate the cascade in the bone morphogenetic pathway. This pathway comprises of BMP proteins and smads, which are transcriptional regulators. These transcriptional regulators are the proteins responsible for downstream changes in gene expression by going into the nucleus of the activated cells and modifying the transcriptional activity of genes. It is this transcriptional change that results in the cellular differentiation of the cell into osteoblasts. The ACVR1 inhibitor FKBP1A also was featured on STRING. The description of the inhibitor is consistent with the literature in indicating that this inhibitor blocks ACVR1 activity by binding to the TGF-beta domain and causing the ACVR1 to be in its inactive conformation in the absence of the ligand. Considering the pathway appears to be constitutively active in the pathological state, having the mutation in the TGF-beta domain disrupts the affinity of the inhibitor to ACVR1, causing leaky signaling of these downstream transcriptional activation changes. Not surprisingly, all this information was also present on STRING. Therefore, data bases like STRING incorporate a vast amount of information that can be useful when determining the interactions into proteins and the biological activity of that protein.
Studies have confirmed that a missense mutation the 206th amino acid in the ACVR1 protein is consistently correlated with FOP. Research has also confirmed that ACVR1 is a transmembrane protein that receives signals from the outside of the cell and facilitates internal cellular changes that ultimately alter gene expression. Therefore, from investigating the information derived from databases like STRING, these interactions confirm what is known from the literature. Using other databases such as PFAM and SMART, domains in ACVR1 were identified and conclusions into protein activity can be inferred from the outputs. As mentioned above, these databases predicted that ACVR1 has kinase ability which confers the protein to stimulate cell signaling upon ligand binding. This information is also present in the literature. Therefore, activation of this protein has enzymatic activity to initiate the cascade in the bone morphogenetic pathway. This pathway comprises of BMP proteins and smads, which are transcriptional regulators. These transcriptional regulators are the proteins responsible for downstream changes in gene expression by going into the nucleus of the activated cells and modifying the transcriptional activity of genes. It is this transcriptional change that results in the cellular differentiation of the cell into osteoblasts. The ACVR1 inhibitor FKBP1A also was featured on STRING. The description of the inhibitor is consistent with the literature in indicating that this inhibitor blocks ACVR1 activity by binding to the TGF-beta domain and causing the ACVR1 to be in its inactive conformation in the absence of the ligand. Considering the pathway appears to be constitutively active in the pathological state, having the mutation in the TGF-beta domain disrupts the affinity of the inhibitor to ACVR1, causing leaky signaling of these downstream transcriptional activation changes. Not surprisingly, all this information was also present on STRING. Therefore, data bases like STRING incorporate a vast amount of information that can be useful when determining the interactions into proteins and the biological activity of that protein.
|
Proteomics
What are post-translational modifications? The number of genes in the genome in humans is estimated by scientist to be 20,000-25,000 (11). Despite the limited number of genes in a human, it is estimated that the number of different proteins in a person is near 1 million (11). Therefore, this brings into question what is allowing for a gene to encode more than one protein? The answer is that a few cellular processes and mechanisms exist that enable one gene to code for multiple proteins, one of them being post-translational modifications (PTM). Figure 5 illustrates how PTMS provide an increase in the complexity of the proteome. PTMS are chemical modifications that can alter many aspects of a protein such as regulating activity, localization, and cellular functions of proteins. Moreover, these PTMS greatly diversify the functions of proteins in the proteome by the covalent addition of these modifications. There are a variety of different types of most translational modifications. Some of the common modifications to proteins include phosphorylation, methylation, acetylation, ubiquination, and glycosylation. These modifications are an integral aspect for studying proteins under normal cell conditions and in pathogenesis by providing clues into understanding cellular activity/processes, disease mechanisms, and disease treatment and prevention. |
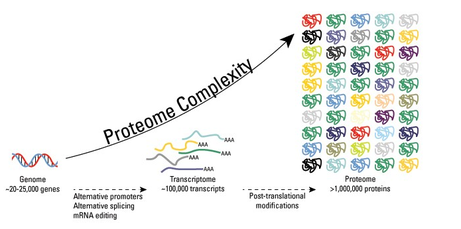
Figure 5: Post translational modifications greatly increase proteomic diversity relative to the transcriptome and genome.
|
What are the phosphorylation sites on ACVR1?
Using sites such as uniprot, expasy, and Netphos information can be derived about protein localization in a cell, protein-protein or binary interactions, tryspin cuts, isoelectric point, and post-translational modifications. Information derived from these databases are listed below:
Uniprot
This database provides a plethora of information about a particular protein of interest. This website provided information on the function of ACVR1 and localization. Specifically, uniprot specified that this is protein is a ligand binding transmembrane receptor composed of two parts, type 1 and type 2. This receptor has serine/threonine kinase activity. This information was used later to investigate the PTM on these catalytic kinase domains. Information from this website indicates that the activation of the protein activates SMAD proteins, which is a transcriptional regulator. This database also indicates the tissue specificity of this protein. Tissues that incorporate ACVR1 proteins include normal parachymal cells, endothelial cells, fibroblasts, and tumor derived epithelial cells. This information can be used for further study into the cells responsible for the transition to bone cells. Currently, it is unclear which cell type is responsible for undergoing the metamorphosis, thus, it would be interesting to investigate which cell type that possesses ACVR1 signaling is responsible for migrating to soft tissue sites and transforming into bone cells. Lastly, another notable feature on this website is that it has links to the interactions ACVR1 makes with other proteins. Links to the publications that discovered these interactions is also provided. For FOP, the protein interactions portion of Uniprot confirm that ACVR1 interacts with FKBP1A from a publication using x-ray crystallography (11).
Expasy
This website can connect the user to a variety of different resources to study a protein of interest. For ACVR1, Expasy was used to determine the cuts made in the protein sequence by various enzymes. Figure 5 below depicts the cuts made in the peptide. Trypsin cuts at arginine or lysine amino acids. hese fragments should be detected using mass spectrometry methods.
Using sites such as uniprot, expasy, and Netphos information can be derived about protein localization in a cell, protein-protein or binary interactions, tryspin cuts, isoelectric point, and post-translational modifications. Information derived from these databases are listed below:
Uniprot
This database provides a plethora of information about a particular protein of interest. This website provided information on the function of ACVR1 and localization. Specifically, uniprot specified that this is protein is a ligand binding transmembrane receptor composed of two parts, type 1 and type 2. This receptor has serine/threonine kinase activity. This information was used later to investigate the PTM on these catalytic kinase domains. Information from this website indicates that the activation of the protein activates SMAD proteins, which is a transcriptional regulator. This database also indicates the tissue specificity of this protein. Tissues that incorporate ACVR1 proteins include normal parachymal cells, endothelial cells, fibroblasts, and tumor derived epithelial cells. This information can be used for further study into the cells responsible for the transition to bone cells. Currently, it is unclear which cell type is responsible for undergoing the metamorphosis, thus, it would be interesting to investigate which cell type that possesses ACVR1 signaling is responsible for migrating to soft tissue sites and transforming into bone cells. Lastly, another notable feature on this website is that it has links to the interactions ACVR1 makes with other proteins. Links to the publications that discovered these interactions is also provided. For FOP, the protein interactions portion of Uniprot confirm that ACVR1 interacts with FKBP1A from a publication using x-ray crystallography (11).
Expasy
This website can connect the user to a variety of different resources to study a protein of interest. For ACVR1, Expasy was used to determine the cuts made in the protein sequence by various enzymes. Figure 5 below depicts the cuts made in the peptide. Trypsin cuts at arginine or lysine amino acids. hese fragments should be detected using mass spectrometry methods.

NetPhos
Netphos is a bioinformatics tool that can be used to predict the phosphorylation post translational modifications on a protein. Netphos was used to predict the phosphorylation sites on ACVR1. After supplying Netphos with the protein sequence of ACVR1, it generates an output such as the document below. Figure 6 below illustrates the phosphorylation sites predicted on the human ACVR1. Netphos provides a value between 0-1 that predicts the liklehood of a phosphorylation site being on a particular amino acid. The colors of the line correlate with amino acids that are predicted to be phosphorylated. The height of the lines correlates with the certainty that these sites are indeed sites of phosphorylation. The higher the bar, the more likely that this amino acid is phosphorylated.
Netphos is a bioinformatics tool that can be used to predict the phosphorylation post translational modifications on a protein. Netphos was used to predict the phosphorylation sites on ACVR1. After supplying Netphos with the protein sequence of ACVR1, it generates an output such as the document below. Figure 6 below illustrates the phosphorylation sites predicted on the human ACVR1. Netphos provides a value between 0-1 that predicts the liklehood of a phosphorylation site being on a particular amino acid. The colors of the line correlate with amino acids that are predicted to be phosphorylated. The height of the lines correlates with the certainty that these sites are indeed sites of phosphorylation. The higher the bar, the more likely that this amino acid is phosphorylated.
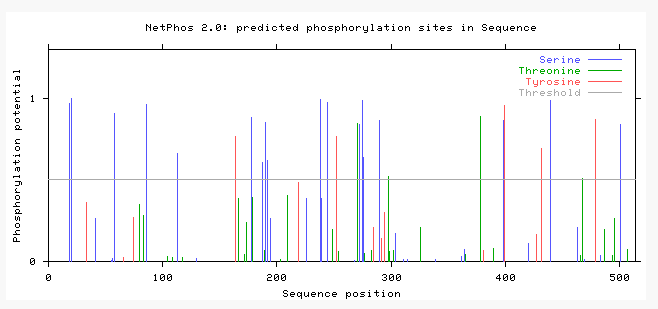
Figure 6: This image represents the predicted phosphorylation sites on ACVR1 in humans. Considering ACVR1 is considered a serine/threonine kinase, there are an abundance of phsophorylations on serines and threonines.
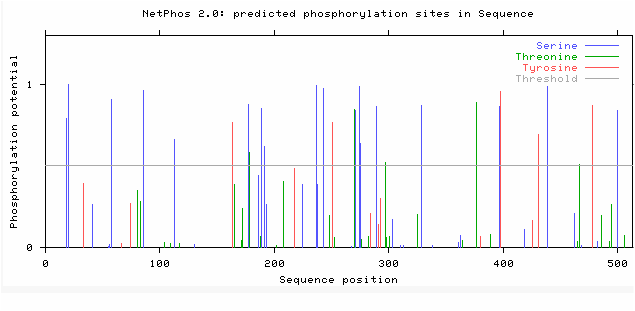
Figure 7: This image represents the phosphorylations in mouse ACVR1. This illustrates that the phosphorylation sites are extremely similar to the human phosphorylation sites.
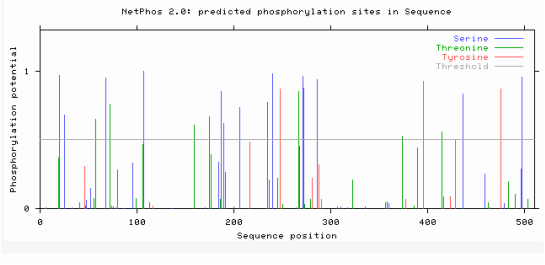
Figure 8: This image represents the phosphorylation sites in the zebrafish ACVR1 sequence.
Analysis:
A few conclusions can be drawn from these bioinformatics tools. First of all, in regards to Expasy, various enzymes can be used to cut the protein of interest into fragments. For aim 3 on the “Conclusions and Future Directions” page, that aim focuses on using an enzyme called trypsin to cut ACVR1 into fragments so it can be read by mass spectrometer. Expasy is been enlightening because it illustrates that trypsin would not be an ideal enzyme to use for aim 3 given the fact trypsin cuts at arginines and lysines. Normally, at the 206th amino acid in ACVR1, an arginine is the wildtype and conserved amino acid, but in the disease state, that amino acid gets replaced with histidine. Therefore, this may not be an ideal enzyme because the peptide lengths are different in the disease state verses the normal peptide. In the normal state, a cut would be made at the 206th amino acid but in the pathological state no cut would be made. Because aim 3 is evaluating the additions of post translational modifications in the disease state vs the normal state, ideally the size of the fragments would be the same and the additions of post translational modifications would be the only component responsible for different fragment lengths. Therefore, I would use a different enzyme such as LysC or LysN.
Secondly, Netphos gives indications as to phosphate additions on proteins. Since ACVR1 is a transmembrane serine/threonine kinase, it is not surprising that there are many phosphorylation sites in this protein. Most notably, the kinase domain has an abundance of phosphorylation sites. Considering this domain is responsible for the phosphorylations of the activation of downstream targets in the BMP pathway, it is not surprising that the kinase domain has many phosphorylation sites. Additionally, the information from NetPhos also suggests that the phosphorylation sites are relatively conserved throughout species. Since human and mice protein sequences are more similar identity, it makes sense that they would have more phosphorylation sites conserved than the zebrafish. I would expect these to be relatively conserved because the BMP pathway is a highly conserved pathway through a variety of species and is crucial in development of vertebrates.
A few conclusions can be drawn from these bioinformatics tools. First of all, in regards to Expasy, various enzymes can be used to cut the protein of interest into fragments. For aim 3 on the “Conclusions and Future Directions” page, that aim focuses on using an enzyme called trypsin to cut ACVR1 into fragments so it can be read by mass spectrometer. Expasy is been enlightening because it illustrates that trypsin would not be an ideal enzyme to use for aim 3 given the fact trypsin cuts at arginines and lysines. Normally, at the 206th amino acid in ACVR1, an arginine is the wildtype and conserved amino acid, but in the disease state, that amino acid gets replaced with histidine. Therefore, this may not be an ideal enzyme because the peptide lengths are different in the disease state verses the normal peptide. In the normal state, a cut would be made at the 206th amino acid but in the pathological state no cut would be made. Because aim 3 is evaluating the additions of post translational modifications in the disease state vs the normal state, ideally the size of the fragments would be the same and the additions of post translational modifications would be the only component responsible for different fragment lengths. Therefore, I would use a different enzyme such as LysC or LysN.
Secondly, Netphos gives indications as to phosphate additions on proteins. Since ACVR1 is a transmembrane serine/threonine kinase, it is not surprising that there are many phosphorylation sites in this protein. Most notably, the kinase domain has an abundance of phosphorylation sites. Considering this domain is responsible for the phosphorylations of the activation of downstream targets in the BMP pathway, it is not surprising that the kinase domain has many phosphorylation sites. Additionally, the information from NetPhos also suggests that the phosphorylation sites are relatively conserved throughout species. Since human and mice protein sequences are more similar identity, it makes sense that they would have more phosphorylation sites conserved than the zebrafish. I would expect these to be relatively conserved because the BMP pathway is a highly conserved pathway through a variety of species and is crucial in development of vertebrates.
References:
1."Family: Activin_recp (PF01064)." Pfam. EMBL-EBI, n.d. Web. 27 Mar. 2015. <http://pfam.xfam.org//family/PF01064.18>.
2."Protein Motifs: Connectivity Between Secondary Structure Elements."Protein Motifs: Connectivity Between Secondary Structure Elements. N.p., n.d. Web. 27 Mar. 2015. <http://www.proteinstructures.com/Structure/Structure/protein-motifs.html>.
3. "Homepage." SMART. N.p., n.d. Web. 27 Mar. 2015. <http://smart.embl-heidelberg.de/>.
4."Protein Motifs." Protein Motifs. N.p., n.d. Web. 27 Mar. 2015. <http://bioweb.uwlax.edu/genweb/molecular/Seq_Anal/Protein_Motifs/protein_motifs.htm>.
5.Bork, P ; Koonin, Ev. "Protein sequence motifs." Current Opinion In Structural Biology, 1996 Jun, Vol.6(3), pp.366-376. http://www.ncbi.nlm.nih.gov/pubmed/8804823
6. "What Are Protein Domains." EMBL-EBI. EMBL-EBI, n.d. Web. 27 Mar. 2015. <http://www.ebi.ac.uk/training/online/course/introduction-protein-classification-ebi/protein-classification/what-are-protein-domains>.
7. "Fibrodysplasia Ossificans Progressiva." Genetics Home Reference. U.S. National Library of Medicine, 9 Feb. 2015. Web. 11 Feb. 2015. http://ghr.nlm.nih.gov/condition/fibrodysplasia-ossificans-progressiva
8. Chen, Y G ; Liu, F ; Massague, J. "Mechanism of TGFbeta receptor inhibition by FKBP12." The EMBO journal, 1 July 1997, Vol.16 (13), pp.3866-76. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1170011/
9. Chaikuad, A ; Alfano, I ; Kerr, G ; Sanvitale, CE ; Boergermann, Jh ; Triffitt, Jt ; Von Delft, F ; Knapp, S ; Knaus, P ; Bullock, An. "Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva." Journal Of Biological Chemistry, 2012 Oct 26, Vol.287(44), pp.36990-36998. http://www.ncbi.nlm.nih.gov/pubmed/22977237
10. Yu, Pb ; Deng, Dy ; Lai, CS ; Hong, Cc ; Cuny, Gd ; Bouxsein, Ml ; Hong, Dw ; Mcmanus, Pm ; Katagiri, T ; Sachidanandan, C ; Kamiya, N ; Fukuda, T ; Mishina, Y ; Peterson, Rt ; Bloch, Kd. "BMP type I receptor inhibition reduces heterotopic ossification." Nature Medicine, 2008 Dec, Vol.14(12), pp.1363-1369. http://www.ncbi.nlm.nih.gov/pubmed/19029982
11. SQSTM - Human. UniProt Consortium. http://www.uniprot.org
Image References:
1. http://pfam.xfam.org
3-4. http://string-db.org/newstring_cgi/show_network_section.pl
5. https://www.lifetechnologies.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-post-translational-modification.html
6-8. http://www.expasy.org
1."Family: Activin_recp (PF01064)." Pfam. EMBL-EBI, n.d. Web. 27 Mar. 2015. <http://pfam.xfam.org//family/PF01064.18>.
2."Protein Motifs: Connectivity Between Secondary Structure Elements."Protein Motifs: Connectivity Between Secondary Structure Elements. N.p., n.d. Web. 27 Mar. 2015. <http://www.proteinstructures.com/Structure/Structure/protein-motifs.html>.
3. "Homepage." SMART. N.p., n.d. Web. 27 Mar. 2015. <http://smart.embl-heidelberg.de/>.
4."Protein Motifs." Protein Motifs. N.p., n.d. Web. 27 Mar. 2015. <http://bioweb.uwlax.edu/genweb/molecular/Seq_Anal/Protein_Motifs/protein_motifs.htm>.
5.Bork, P ; Koonin, Ev. "Protein sequence motifs." Current Opinion In Structural Biology, 1996 Jun, Vol.6(3), pp.366-376. http://www.ncbi.nlm.nih.gov/pubmed/8804823
6. "What Are Protein Domains." EMBL-EBI. EMBL-EBI, n.d. Web. 27 Mar. 2015. <http://www.ebi.ac.uk/training/online/course/introduction-protein-classification-ebi/protein-classification/what-are-protein-domains>.
7. "Fibrodysplasia Ossificans Progressiva." Genetics Home Reference. U.S. National Library of Medicine, 9 Feb. 2015. Web. 11 Feb. 2015. http://ghr.nlm.nih.gov/condition/fibrodysplasia-ossificans-progressiva
8. Chen, Y G ; Liu, F ; Massague, J. "Mechanism of TGFbeta receptor inhibition by FKBP12." The EMBO journal, 1 July 1997, Vol.16 (13), pp.3866-76. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1170011/
9. Chaikuad, A ; Alfano, I ; Kerr, G ; Sanvitale, CE ; Boergermann, Jh ; Triffitt, Jt ; Von Delft, F ; Knapp, S ; Knaus, P ; Bullock, An. "Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva." Journal Of Biological Chemistry, 2012 Oct 26, Vol.287(44), pp.36990-36998. http://www.ncbi.nlm.nih.gov/pubmed/22977237
10. Yu, Pb ; Deng, Dy ; Lai, CS ; Hong, Cc ; Cuny, Gd ; Bouxsein, Ml ; Hong, Dw ; Mcmanus, Pm ; Katagiri, T ; Sachidanandan, C ; Kamiya, N ; Fukuda, T ; Mishina, Y ; Peterson, Rt ; Bloch, Kd. "BMP type I receptor inhibition reduces heterotopic ossification." Nature Medicine, 2008 Dec, Vol.14(12), pp.1363-1369. http://www.ncbi.nlm.nih.gov/pubmed/19029982
11. SQSTM - Human. UniProt Consortium. http://www.uniprot.org
Image References:
1. http://pfam.xfam.org
3-4. http://string-db.org/newstring_cgi/show_network_section.pl
5. https://www.lifetechnologies.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-post-translational-modification.html
6-8. http://www.expasy.org
