This web page was produced as an assignment for genetics 564, an undergraduate course at UW-Madison.
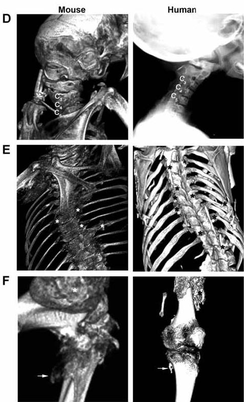 Image 1: This demonstrates the similarities between a FOP mouse model with the phenotype found in human patients of FOP. (Chakkalakal 2012)
Image 1: This demonstrates the similarities between a FOP mouse model with the phenotype found in human patients of FOP. (Chakkalakal 2012)
Introduction
Fibrodysplasia Ossificans Progressiva is a disease that causes excessive and irreversible bone formation at soft tissue sites, consequently, causing an overgrowth of bone over the skeleton (1). This overgrowth of bone eventually results in reduced mobility. FOP is caused by a single missense mutation in the 206th amino acid (R206H) within a transmembrane protein called activin A receptor type I (ACVR1) (1). The mutation resides in the TGF-β domain of the ACVR1 protein. TGF- β domains are important for controlling proliferation and differentiation among other things; therefore, this domain is crucial for normal protein function (2). FOP is thought to be caused by constitutive activation of the bone morphogenetic pathway by the mutated TGF- β domain in the ACVR1 gene, leading to excessive signaling that promotes bone differentiation. Currently, there is no cure. However, the hope is that with further research, a treatment will be on the horizon. Before treatments can be ensued, further research to delineate the molecular mechanisms in FOP should be performed to better understand potential therapeutic approaches.
Zebrafish and mice have been used extensively as models to study FOP (3). More specifically, the mouse ACVR1 homolog protein is 98% identical to the human protein which makes it a great candidate for study (4). This striking similarity is important because a knock in mouse line can be created to mimic similar phenotypes to the human FOP disease state (5). A mouse knock-in was created with the exact same mutation as in the human ACVR1 (R206H) and is shown in image 1 (5). Additionally, zebrafish have been useful in modeling FOP because they do have phenotypes associated with FOP. For these experiments, because FOP is a dominantly inherited disorder, only one pathogenic copy of the ACVR1 gene is needed to obtain a disease phenotype, therefore, heterozygous models will be used.
Aim 1
The focus of aim 1 is to identifying differential gene expression between a wild type mouse and a heterozygous mouse for FOP. A heterozygous mouse will be used since one mutant copy is sufficient for FOP onset. This assay will be performed by taking damaged muscle tissue from a FOP mouse and wild type mouse and comparing the transcriptome using RNA seq. The hope is that this analysis will specify the molecular mechanisms involved in producing the FOP phenotype i.e. soft tissue transforming into bone.
The expected experimental results are shown in image 2. In the literature, researchers hypothesize that the mutation in the TGF- β domain results in decreased binding affinity of the inhibitor FKBP12 for ACVR1 (6,7). The decreased binding affinity of the ACVR1 inhibitor results in ACVR1 being active at inappropriate times and causes changes in downstream signaling and gene expression changes. The downstream signaling is in the bone morphogenetic pathway. Therefore, I would expect in the normal wildtype case that there is a normal default level of the proteins that interact with ACVR1, namely the smad transcription factors and BMP proteins. In the pathological state, since the phenotype suggests this pathway is over stimulated, I would expect the transcription level of genes that produce smads and BMPs to be activated. This hypothesis is depicted in image 2 below. As shown from the STRING database, it is well established in the literature that smads and BMP proteins interact with ACVR1, however, I would also expect in addition to these proteins, other transcription factors associated with osteoblast differentiation are also unregulated in the pathological state (8). Image 3 specifies some additional transcription factors that are an integral element in osteoblast differentiation (9). In FOP, mesenchyme cells that reside in the soft tissue are re-differentiating into osteoblasts. Although the exact mechanisms that result in the transformation of soft tissue to bone are unresolved, it is likely that mesenchyme cells in the soft tissue that adopt an osteoblast fate may be expressing these transcription factors at a higher level than the wildtype. Overall, this experiment achieves insight into the deferentially expressed genes which is important in developing targets for therapeutic methods in the long term.
Fibrodysplasia Ossificans Progressiva is a disease that causes excessive and irreversible bone formation at soft tissue sites, consequently, causing an overgrowth of bone over the skeleton (1). This overgrowth of bone eventually results in reduced mobility. FOP is caused by a single missense mutation in the 206th amino acid (R206H) within a transmembrane protein called activin A receptor type I (ACVR1) (1). The mutation resides in the TGF-β domain of the ACVR1 protein. TGF- β domains are important for controlling proliferation and differentiation among other things; therefore, this domain is crucial for normal protein function (2). FOP is thought to be caused by constitutive activation of the bone morphogenetic pathway by the mutated TGF- β domain in the ACVR1 gene, leading to excessive signaling that promotes bone differentiation. Currently, there is no cure. However, the hope is that with further research, a treatment will be on the horizon. Before treatments can be ensued, further research to delineate the molecular mechanisms in FOP should be performed to better understand potential therapeutic approaches.
Zebrafish and mice have been used extensively as models to study FOP (3). More specifically, the mouse ACVR1 homolog protein is 98% identical to the human protein which makes it a great candidate for study (4). This striking similarity is important because a knock in mouse line can be created to mimic similar phenotypes to the human FOP disease state (5). A mouse knock-in was created with the exact same mutation as in the human ACVR1 (R206H) and is shown in image 1 (5). Additionally, zebrafish have been useful in modeling FOP because they do have phenotypes associated with FOP. For these experiments, because FOP is a dominantly inherited disorder, only one pathogenic copy of the ACVR1 gene is needed to obtain a disease phenotype, therefore, heterozygous models will be used.
Aim 1
The focus of aim 1 is to identifying differential gene expression between a wild type mouse and a heterozygous mouse for FOP. A heterozygous mouse will be used since one mutant copy is sufficient for FOP onset. This assay will be performed by taking damaged muscle tissue from a FOP mouse and wild type mouse and comparing the transcriptome using RNA seq. The hope is that this analysis will specify the molecular mechanisms involved in producing the FOP phenotype i.e. soft tissue transforming into bone.
The expected experimental results are shown in image 2. In the literature, researchers hypothesize that the mutation in the TGF- β domain results in decreased binding affinity of the inhibitor FKBP12 for ACVR1 (6,7). The decreased binding affinity of the ACVR1 inhibitor results in ACVR1 being active at inappropriate times and causes changes in downstream signaling and gene expression changes. The downstream signaling is in the bone morphogenetic pathway. Therefore, I would expect in the normal wildtype case that there is a normal default level of the proteins that interact with ACVR1, namely the smad transcription factors and BMP proteins. In the pathological state, since the phenotype suggests this pathway is over stimulated, I would expect the transcription level of genes that produce smads and BMPs to be activated. This hypothesis is depicted in image 2 below. As shown from the STRING database, it is well established in the literature that smads and BMP proteins interact with ACVR1, however, I would also expect in addition to these proteins, other transcription factors associated with osteoblast differentiation are also unregulated in the pathological state (8). Image 3 specifies some additional transcription factors that are an integral element in osteoblast differentiation (9). In FOP, mesenchyme cells that reside in the soft tissue are re-differentiating into osteoblasts. Although the exact mechanisms that result in the transformation of soft tissue to bone are unresolved, it is likely that mesenchyme cells in the soft tissue that adopt an osteoblast fate may be expressing these transcription factors at a higher level than the wildtype. Overall, this experiment achieves insight into the deferentially expressed genes which is important in developing targets for therapeutic methods in the long term.
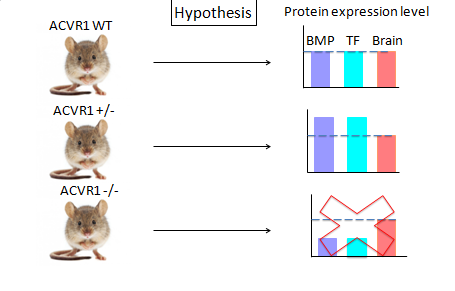
Image 2: This illustrates the hypothesis from Aim 1. This indicates that it is likely that smads and BMP genes have increased expression in the disease state compared to the wildtype.
|
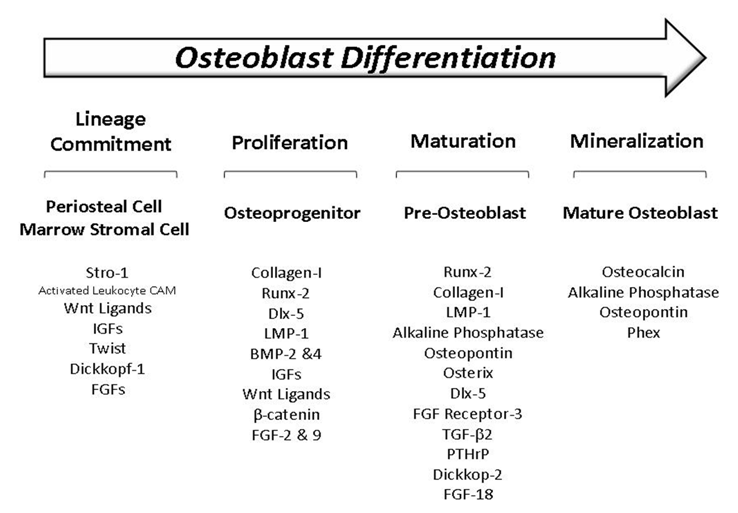
Image 3: This table summarizes important transcription factors involved in osteoblast differentiation. It is likely that these are also over expressed in the disease state.
|
Aim 2
The purpose of Aim 2 is to identify potential drug targets for ACVR1. Using PubChem, the hope is that using the drugs that act as inhibitors on ACVR1 will rescue the aberrant signaling and prevent the bone formation (10). The setup of this experiment consists of using wildtype and heterozygous mutant zebra fish. Since it is believed that in FOP the ACVR1 protein has kinase activity and is phosphorylating downstream targets, it is hypothesized that by adding the inhibitors specific to ACVR1, that it would prevent the initiation of downstream signaling and BMP activation. The experimental apparatus consists of mutant zebrafishes being placed in a well of a 96 well plate. Each zebra fish will receive a different inhibitor compound from Pubchem. The zebra fishes will then receive a needle prick to induce tissue damage similar to a vaccine that initiates flare-ups in humans. The hope is that if any of these inhibitors work, that they would prevent ACVR1 signaling and undergo normal tissue repair mechanisms instead of excessive bone growth. For the wildtype zebra fish, since it is believed in normal conditions that the BMP pathway is not activated upon tissue damage, by adding activator drugs for ACVR1, the disease state can be simulated in the wildtype. Subsequently, a needle prick would follow up exposure to activator drugs. If the drugs worked and stimulated ACVR1 signaling, it can be hypothesized that the wildtype zebra fish would undergo bone differentiation at the damaged site to mimic the disease state. Overall, the hope is a drug to limit the activation of the ACVR1 will rescue the disease phenotype associated with the over stimulation of this pathway. Image 4 details the process of this experiment.
The purpose of Aim 2 is to identify potential drug targets for ACVR1. Using PubChem, the hope is that using the drugs that act as inhibitors on ACVR1 will rescue the aberrant signaling and prevent the bone formation (10). The setup of this experiment consists of using wildtype and heterozygous mutant zebra fish. Since it is believed that in FOP the ACVR1 protein has kinase activity and is phosphorylating downstream targets, it is hypothesized that by adding the inhibitors specific to ACVR1, that it would prevent the initiation of downstream signaling and BMP activation. The experimental apparatus consists of mutant zebrafishes being placed in a well of a 96 well plate. Each zebra fish will receive a different inhibitor compound from Pubchem. The zebra fishes will then receive a needle prick to induce tissue damage similar to a vaccine that initiates flare-ups in humans. The hope is that if any of these inhibitors work, that they would prevent ACVR1 signaling and undergo normal tissue repair mechanisms instead of excessive bone growth. For the wildtype zebra fish, since it is believed in normal conditions that the BMP pathway is not activated upon tissue damage, by adding activator drugs for ACVR1, the disease state can be simulated in the wildtype. Subsequently, a needle prick would follow up exposure to activator drugs. If the drugs worked and stimulated ACVR1 signaling, it can be hypothesized that the wildtype zebra fish would undergo bone differentiation at the damaged site to mimic the disease state. Overall, the hope is a drug to limit the activation of the ACVR1 will rescue the disease phenotype associated with the over stimulation of this pathway. Image 4 details the process of this experiment.
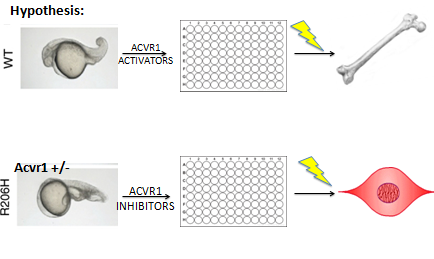
Image 4: This summarizes how the assay for aim 2 will be performed and what the expected result would be.
|
Aim 3
In the literature, it is well established that the FKBP12 inhibitor likely has decreased binding affinity for ACVR1, which is what makes ACVR1 constitutively active in the disease state (6,7). The question becomes what is it that makes this inhibitor have decreased binding affinity for ACVR1? The literature seems to suggest from protein modeling studies that there is a misfolding of the TGF- β domain with the substitute of histidine amino acid for arginine at the 206th location in the protein peptide. However, no proteomic or genomic experiments have been done to confirm this. It turns out that in about 6% of phosphorylations in a cell can occur on histidines in addition to tyrosines, serines, and threonines (11). For this aim, the purpose is to determine if this missense mutation in the 206th amino acid results in a new phosphorylation site that leads to constitutive activation of ACVR1 and decreased binding affinity of FKBP12. In order to confirm this, Expasy will be |
used to identify the fragment size that contains the missense mutation in the ACVR1 peptide after receiving a hypothetical LysC cut in the ACVR1 peptide (12). The fragmented proteins will then be placed in HPLC-MS/MS to evaluate the fragments of ACVR1. I would hypothesize that if the mutation in the disease state did indeed form an additional phosphorylation site, that there would be a shift in fragment weight on the mass spectrometry readout to account for the additional phosphorylation. However, if there is no shift between the control and the FOP mouse sample, then this confirms that perhaps the FKBP12 inhibitor has decreased affinity for ACVR1 due to misfoldings in the domain. The rationale behind this experiment would be to uncover the exact mechanisms that are responsible for decreased inhibitor binding which is responsible for disease phenotype onset.
Future directions
FOP is caused by a single missense mutation in the ACVR1 protein, an essential transmembrane protein involved in initiating signaling in a highly conserved pathway, the BMP pathway (6). This pathway is essential for bone to be formed from cartilage. The exact mechanisms are still being deciphered as to how exactly soft tissue damage triggers this metamorphosis into bone. Once this mystery is solved, that will likely allow for further exploration into treatments for FOP. In addition to unlocking how this pathway gets activated in the first place, another crucial direction for research includes discovering ways to divert the mesenchyme cells in the damaged tissue from adopting an osteoblast fate and more of a soft tissue fate. As mentioned above, since it is believed that ACVR1 is active, a promising avenue to explore for treatments involves finding drugs that will diminish the aberrant signaling of ACVR1, leading to bone gene expression. Lastly, the immune response is an integral component in inducing flare-ups of bone formation upon damage. Therefore, it would be beneficial to discover ways to decrease the inflammatory response. Decreasing the inflammatory response may reduce the severity of bone growth and discomfort associated with tissue damage.
Future directions
FOP is caused by a single missense mutation in the ACVR1 protein, an essential transmembrane protein involved in initiating signaling in a highly conserved pathway, the BMP pathway (6). This pathway is essential for bone to be formed from cartilage. The exact mechanisms are still being deciphered as to how exactly soft tissue damage triggers this metamorphosis into bone. Once this mystery is solved, that will likely allow for further exploration into treatments for FOP. In addition to unlocking how this pathway gets activated in the first place, another crucial direction for research includes discovering ways to divert the mesenchyme cells in the damaged tissue from adopting an osteoblast fate and more of a soft tissue fate. As mentioned above, since it is believed that ACVR1 is active, a promising avenue to explore for treatments involves finding drugs that will diminish the aberrant signaling of ACVR1, leading to bone gene expression. Lastly, the immune response is an integral component in inducing flare-ups of bone formation upon damage. Therefore, it would be beneficial to discover ways to decrease the inflammatory response. Decreasing the inflammatory response may reduce the severity of bone growth and discomfort associated with tissue damage.
| heilingoetterfinalpresentation5-12-15.pdf |
Citations:
1. Kaplan, F. S., Xu, M., Seemann, P., Connor, J. M., Glaser, D. L., Carroll, L., Delai, P., Fastnacht-Urban, E., Forman, S. J., GillessenKaesbach, G., Hoover-Fong, J., Koster, B., Pauli, R. M., Reardon, W., Zaidi, S. A., Zasloff, M., Morhart, R., Mundlos, S., Groppe, J., and Shore, E. M. (2009) Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1, Hum. Mutat. 30, 379–390. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2921861/
2. "Homepage." SMART. N.p., n.d. Web. 27 Mar. 2015. <http://smart.embl-heidelberg.de/>.
3. Shen, Q ; Little, SC ; Xu, Mq ; Haupt, J ; Ast, C ; Katagiri, T ; Mundlos, S ; Seemann, P ; Kaplan, Fs ; Mullins, MC ; Shore, Em.
The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. Journal Of Clinical Investigation, 2009 Nov, Vol.119(11), pp.3462-3472. http://www.ncbi.nlm.nih.gov/pubmed/19855136
4. "Discover Homologs." National Center for Biotechnology Information. U.S. National Library of Medicine, n.d. Web. 27 Mar. 2015. <http://www.ncbi.nlm.nih.gov/homologene>.
5. Chakkalakal, S. A., Zhang, D., Culbert, A. L., Convente, M. R., Caron, R. J., Wright, A. C., Maidment, A. D. A., Kaplan, F. S. and Shore, E. M. (2012). An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J. Bone Miner. Res. 27, 1746-1756.. 5. "Fibrodysplasia Ossificans Progressiva." Genetics Home Reference. U.S. National Library of Medicine, 9 Feb. 2015. Web. 11 Feb. 2015. http://onlinelibrary.wiley.com.ezproxy.library.wisc.edu/doi/10.1002/jbmr.1637/pdf
6. Chaikuad, A ; Alfano, I ; Kerr, G ; Sanvitale, CE ; Boergermann, Jh ; Triffitt, Jt ; Von Delft, F ; Knapp, S ; Knaus, P ; Bullock, An. Structure of the Bone Morphogenetic Protein Receptor ALK2 and Implications for Fibrodysplasia OssificansProgressiva. Journal Of Biological Chemistry, 2012 Oct 26, Vol.287(44), pp.36990-36998
7. Chen, Y G ; Liu, F ; Massague, J. Mechanism of TGFbeta receptor inhibition by FKBP12. The EMBO journal, 1 1997, Vol.16(13), pp.3866-76. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1170011/
8. http://string-db.org/
9. Strohbach, Cassandra ; Rundle, Charles ; Wergedal, Jon ; Chen, Shin-Tai ; Linkhart, Thomas ; Lau, K.-H. ; Strong, Donna. Gene Therapy Applications for Fracture Repair. Calcified Tissue International, 2008, Vol.83(3), pp.202-211. http://www.intechopen.com/books/gene-therapy-applications/gene-therapy-applications-for-fracture-repair
10. https://pubchem.ncbi.nlm.nih.gov/search/
11. Puttick, J ; Baker, En ; Delbaere, Ltj. Histidine phosphorylation in biological systems. Biochimica Et Biophysica Acta-Proteins And Proteomics, 2008 Jan, Vol.1784(1), pp.100 105. http://www.sciencedirect.com/science/article/pii/S1570963907001586
12. http://www.expasy.org/
13. Lowery, JW ; Rosen, V. Allele-Specific RNA Interference in FOPSilencing the FOP gene. Gene Therapy, 2012 Jul, Vol.19(7), pp.701-702. http://www.nature.com/gt/journal/v19/n7/full/gt2011190a.html
14. Lounev, Vy ; Ramachandran, R ; Wosczyna, Mn ; Yamamoto, M ; Maidment, ADA ; Shore, Em ; Glaser, DL ; Goldhamer, Dj ; Kaplan, Fs. Identification of Progenitor Cells That Contribute to Heterotopic Skeletogenesis. Journal Of Bone And Joint Surgery-American Volume, 2009 Mar(3), pp.652-663. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2663346/
15.Chaikuad, A ; Alfano, I ; Kerr, G ; Sanvitale, CE ; Boergermann, Jh ; Triffitt, Jt ; Von Delft, F ; Knapp, S ; Knaus, P ; Bullock, An. Structure of the Bone Morphogenetic Protein Receptor ALK2 and Implications for Fibrodysplasia OssificansProgressiva. Journal Of Biological Chemistry, 2012 Oct 26, Vol.287(44), pp.36990-36998
1. Kaplan, F. S., Xu, M., Seemann, P., Connor, J. M., Glaser, D. L., Carroll, L., Delai, P., Fastnacht-Urban, E., Forman, S. J., GillessenKaesbach, G., Hoover-Fong, J., Koster, B., Pauli, R. M., Reardon, W., Zaidi, S. A., Zasloff, M., Morhart, R., Mundlos, S., Groppe, J., and Shore, E. M. (2009) Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1, Hum. Mutat. 30, 379–390. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2921861/
2. "Homepage." SMART. N.p., n.d. Web. 27 Mar. 2015. <http://smart.embl-heidelberg.de/>.
3. Shen, Q ; Little, SC ; Xu, Mq ; Haupt, J ; Ast, C ; Katagiri, T ; Mundlos, S ; Seemann, P ; Kaplan, Fs ; Mullins, MC ; Shore, Em.
The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. Journal Of Clinical Investigation, 2009 Nov, Vol.119(11), pp.3462-3472. http://www.ncbi.nlm.nih.gov/pubmed/19855136
4. "Discover Homologs." National Center for Biotechnology Information. U.S. National Library of Medicine, n.d. Web. 27 Mar. 2015. <http://www.ncbi.nlm.nih.gov/homologene>.
5. Chakkalakal, S. A., Zhang, D., Culbert, A. L., Convente, M. R., Caron, R. J., Wright, A. C., Maidment, A. D. A., Kaplan, F. S. and Shore, E. M. (2012). An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J. Bone Miner. Res. 27, 1746-1756.. 5. "Fibrodysplasia Ossificans Progressiva." Genetics Home Reference. U.S. National Library of Medicine, 9 Feb. 2015. Web. 11 Feb. 2015. http://onlinelibrary.wiley.com.ezproxy.library.wisc.edu/doi/10.1002/jbmr.1637/pdf
6. Chaikuad, A ; Alfano, I ; Kerr, G ; Sanvitale, CE ; Boergermann, Jh ; Triffitt, Jt ; Von Delft, F ; Knapp, S ; Knaus, P ; Bullock, An. Structure of the Bone Morphogenetic Protein Receptor ALK2 and Implications for Fibrodysplasia OssificansProgressiva. Journal Of Biological Chemistry, 2012 Oct 26, Vol.287(44), pp.36990-36998
7. Chen, Y G ; Liu, F ; Massague, J. Mechanism of TGFbeta receptor inhibition by FKBP12. The EMBO journal, 1 1997, Vol.16(13), pp.3866-76. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1170011/
8. http://string-db.org/
9. Strohbach, Cassandra ; Rundle, Charles ; Wergedal, Jon ; Chen, Shin-Tai ; Linkhart, Thomas ; Lau, K.-H. ; Strong, Donna. Gene Therapy Applications for Fracture Repair. Calcified Tissue International, 2008, Vol.83(3), pp.202-211. http://www.intechopen.com/books/gene-therapy-applications/gene-therapy-applications-for-fracture-repair
10. https://pubchem.ncbi.nlm.nih.gov/search/
11. Puttick, J ; Baker, En ; Delbaere, Ltj. Histidine phosphorylation in biological systems. Biochimica Et Biophysica Acta-Proteins And Proteomics, 2008 Jan, Vol.1784(1), pp.100 105. http://www.sciencedirect.com/science/article/pii/S1570963907001586
12. http://www.expasy.org/
13. Lowery, JW ; Rosen, V. Allele-Specific RNA Interference in FOPSilencing the FOP gene. Gene Therapy, 2012 Jul, Vol.19(7), pp.701-702. http://www.nature.com/gt/journal/v19/n7/full/gt2011190a.html
14. Lounev, Vy ; Ramachandran, R ; Wosczyna, Mn ; Yamamoto, M ; Maidment, ADA ; Shore, Em ; Glaser, DL ; Goldhamer, Dj ; Kaplan, Fs. Identification of Progenitor Cells That Contribute to Heterotopic Skeletogenesis. Journal Of Bone And Joint Surgery-American Volume, 2009 Mar(3), pp.652-663. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2663346/
15.Chaikuad, A ; Alfano, I ; Kerr, G ; Sanvitale, CE ; Boergermann, Jh ; Triffitt, Jt ; Von Delft, F ; Knapp, S ; Knaus, P ; Bullock, An. Structure of the Bone Morphogenetic Protein Receptor ALK2 and Implications for Fibrodysplasia OssificansProgressiva. Journal Of Biological Chemistry, 2012 Oct 26, Vol.287(44), pp.36990-36998
